Más sobre ciencia de datos en: cienciadedatos.net
- Regresión lineal con Python
- Regresión lineal múltiple con Python
- Regresión logística con Python
- Regularización Ridge, Lasso y Elastic Net con Python
- Machine learning con Python y Scikitlearn
- Árboles de decisión con Python: regresión y clasificación
- Random Forest con Python y Scikit-learn
- Gradient Boosting con Python y Scikit-learn
- Gradient Boosting probabilístico con Python
- Máquinas de Vector Soporte (SVM)
- Redes neuronales con Python
- Análisis de componentes principales PCA
- Clustering
- Detección de anomalías con PCA
- Detección de anomalías con autoencoders
- Detección de anomalías con Gaussian Mixture Models
- Detección de anomalías con Isolation Forest
- Análisis de texto (text mining) con Python
- Reglas de asociación
- Análisis Tweets con Python
Introducción¶
El término clustering hace referencia a un amplio abanico de técnicas cuya finalidad es encontrar patrones o grupos (clusters) dentro de un conjunto de observaciones. Las particiones se establecen de forma que las observaciones que están dentro de un mismo grupo son similares entre ellas y distintas a las observaciones de otros grupos. Se trata de un método de aprendizaje no supervisado (unsupervised), ya que el proceso no tiene en cuenta a qué grupo pertenece realmente cada observación (si es que existe tal información). Esta característica es la que diferencia al clustering de los métodos de clasificación en el que sí emplea la verdadera clasificación durante su entrenamiento.
Dada la utilidad del clustering en disciplinas muy distintas (genómica, marketing...), se han desarrollado multitud de variantes y adaptaciones de sus métodos y algoritmos. Pueden diferenciarse tres grupos principales:
Partitioning Clustering: este tipo de algoritmos requieren que el usuario especifique de antemano el número de clusters que se van a crear (K-means, K-medoids, CLARA).
Hierarchical Clustering: este tipo de algoritmos no requieren que el usuario especifique de antemano el número de clusters. (agglomerative clustering, divisive clustering).
Métodos que combinan o modifican los anteriores (hierarchical K-means, fuzzy clustering, model based clustering y density based clustering).
La librería Scikit Learn contiene implementaciones en Python de los principales algoritmos de clustering.
Medidas de distancia¶
Todos los métodos de clustering tienen una cosa en común, para llevar a cabo las agrupaciones necesitan definir y cuantificar la similitud entre las observaciones. El término distancia se emplea dentro del contexto del clustering como cuantificación de la similitud o diferencia entre observaciones. Si se representan las observaciones en un espacio $p$ dimensional, siendo $p$ el número de variables asociadas a cada observación, cuando más se asemejen dos observaciones más próximas estarán, de ahí que se emplee el término distancia. La característica que hace del clustering un método adaptable a escenarios muy diversos es que puede emplear casi cualquier tipo de distancia, lo que permite al investigador escoger la más adecuada para el estudio en cuestión. A continuación, se describen algunas de las más utilizadas.
Distancia euclídea¶
La distancia euclídea entre dos puntos $p$ y $q$ se define como la longitud del segmento que une ambos puntos. En coordenadas cartesianas, la distancia euclídea se calcula empleando el teorema de Pitágoras. Por ejemplo, en un espacio de dos dimensiones en el que cada punto está definido por las coordenadas $(x,y)$, la distancia euclídea entre $p$ y $q$ es:
$$d_{euc}(p,q) = \sqrt{(p_x - q_x)^2 + (p_y - q_y)^2}$$Esta ecuación puede generalizarse para un espacio euclídeo n-dimensional donde cada punto está definido por un vector de n coordenadas: $p = (p_1, p_2, p_3,...,p_n)$ y $q = (q_1, q_2, q_3,...,q_n)$.
$$d_{euc}(p,q) = \sqrt{(p_1 - q_1)^2 + (p_2 - q_2)^2 +...+(p_n - q_n)^2} =$$$$\sqrt{ \sum_{i=1}^n (p_i - q_i)^2}$$Una forma de dar mayor peso a aquellas observaciones que están más alejadas es emplear la distancia euclídea al cuadrado. En el caso del clustering, donde se busca agrupar observaciones que minimicen la distancia, esto se traduce en una mayor influencia de aquellas observaciones que están más distantes.
Distancia de Manhattan¶
La distancia de Manhattan, también conocida como taxicab metric, rectilinear distance o L1 distance, define la distancia entre dos puntos $p$ y $q$ como el sumatorio de las diferencias absolutas entre cada dimensión. Esta medida se ve menos afectada por outliers (es más robusta) que la distancia euclídea debido a que no eleva al cuadrado las diferencias.
$$d_{man}(p,q) = \sum_{i=1}^n |(p_i - q_i)|$$La siguiente imagen muestra una comparación entre la distancia euclídea (segmento verde) y la distancia de manhattan (segmento rojo, amarillo y azul) en un espacio bidimensional. Existen múltiples caminos para unir dos puntos con una misma distancia de manhattan, pero solo uno con la distancia euclídea.
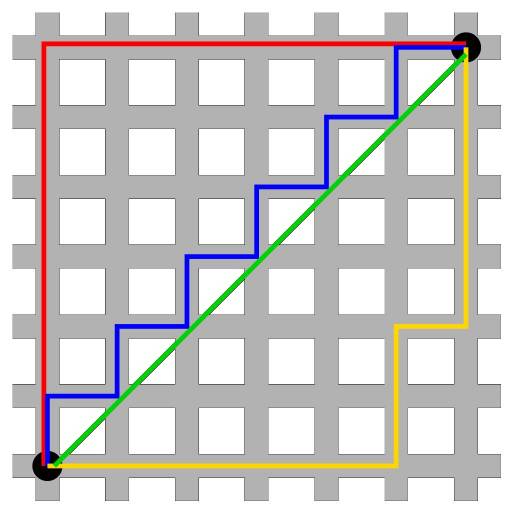
Correlación¶
La correlación es una medida de distancia muy útil cuando la definición de similitud se hace en términos de patrón o forma y no de desplazamiento o magnitud.
$$d_{cor}(p,q) = 1 - \text{correlacion}(p,q)$$donde la correlación puede ser de distintos tipos (Pearson, Spearman, Kendall...)
En la siguiente imagen se muestra el perfil de 3 observaciones. a y b tienen exactamente el mismo patrón, pero b está desplazada 4 unidades por debajo de a. La observación c tiene un patrón distinto a las demás, pero su rango de valores es similar al de b. Acorde a la distancia euclídea, las observaciones b y c son las más similares ya que la suma de la distancia al cuadrado entre sus valores es menor. Sin embargo, acorde a la correlación de Pearson, las observaciones más similares son a y b ya que siguen el mismo patrón.
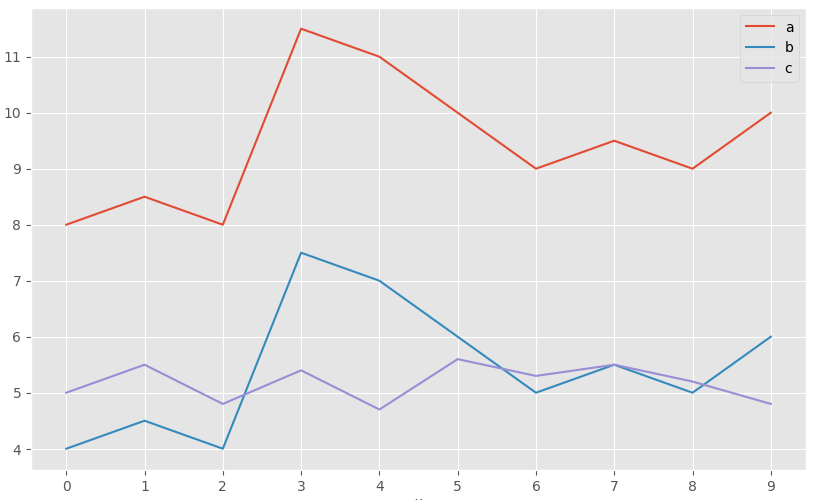
Este ejemplo pone de manifiesto que no existe una única medida de distancia que sea mejor que las demás, sino que depende del contexto en el que se utilice.
Correlación Jackknife¶
El coeficiente de correlación de Pearson resulta efectivo como medida de similitud en ámbitos muy diversos. Sin embargo, tiene la desventaja de no ser robusto frente a outliers, a pesar de que se cumpla la condición de normalidad. Si dos variables tienen un pico o un valle común en una única observación, por ejemplo, por un error de lectura, la correlación va a estar dominada por este registro a pesar de que entre las dos variables no exista correlación real. Lo mismo puede ocurrir en la dirección opuesta. Si dos variables están altamente correlacionadas excepto para una observación, en la que los valores son muy dispares, entonces la correlación existente quedará enmascarada. Una forma de evitarlo es recurrir a la Jackknife correlation, que consiste en calcular todos los posibles coeficientes de correlación entre dos variables si se excluye cada vez una de las observaciones. El promedio de todas las Jackknife correlations atenúa en cierta medida el efecto del outlier.
$$\bar{\theta}_{(A,B)} = \text{Promedio Jackknife correlation (A,B)} = \frac{1}{n} \sum_{i=1}^n \hat r_i$$donde $n$ es el número de observaciones y $\hat r_i$ es el coeficiente de correlación entre las variables $A$ y $B$, habiendo excluido la observación $i$.
Además del promedio, se puede estimar su error estándar ($SE$) y así obtener intervalos de confianza para la Jackknife correlation y su correspondiente p-value.
$$SE = \sqrt{\frac{n-1}{n} \sum_{i=1}^n (\hat r_i - \bar{\theta})^2}$$Intervalo de confianza del 95% $(Z=1.96)$
$$\text{Promedio Jackknife correlation (A,B)} \pm \ 1.96 *SE$$$$\bar{\theta} - 1.96 \sqrt{\frac{n-1}{n} \sum_{i=1}^n (\hat r_i - \bar{\theta})^2} \ , \ \ \bar{\theta}+ 1.96 \sqrt{\frac{n-1}{n} \sum_{i=1}^n (\hat r_i - \bar{\theta})^2}$$P-value para la hipótesis nula de que $\bar{\theta} = 0$:
$$Z_{calculada} = \frac{\bar{\theta} - H_0}{SE}= \frac{\bar{\theta} - 0}{\sqrt{\frac{n-1}{n} \sum_{i=1}^n (\hat r_i - \bar{\theta})^2}}$$$$p_{value} = P(Z > Z_{calculada})$$Cuando se emplea este método, es conveniente calcular la diferencia entre el valor de correlación obtenido por Jackknife correlation $(\bar{\theta})$ y el que se obtiene si se emplean todas las observaciones $(\bar{r})$. A esta diferencia se le conoce como Bias. Su magnitud es un indicativo de cuanto está influenciada la estimación de la correlación entre dos variables debido a un valor atípico u outlier.
$$Bias = (n-1)*(\bar{\theta} - \hat{r})$$Si se calcula la diferencia entre cada correlación $(\hat r_i)$ estimada en el proceso de Jackknife y el valor de correlación $(\hat r)$ obtenido si se emplean todas las observaciones, se puede identificar que observaciones son más influyentes.
Cuando el estudio requiere minimizar al máximo la presencia de falsos positivos, a pesar de que se incremente la de falsos negativos, se puede seleccionar como valor de correlación el menor de entre todos los calculados en el proceso de Jackknife.
$$\text{Correlacion} = \text{min} \{ \hat r_1, \hat r_2,..., \hat r_n \}$$A pesar de que el método de Jackknife permite aumentar la robustez de la correlación de Pearson, si los outliers son muy extremos su influencia seguirá siendo notable.
Simple matching coefficient¶
Cuando las variables con las que se pretende determinar la similitud entre observaciones son de tipo binario, a pesar de que es posible codificarlas de forma numérica como $1$ o $0$, no tiene sentido aplicar operaciones aritméticas sobre ellas (media, suma...). Por ejemplo, si la variable sexo se codifica como $1$ para mujer y $0$ para hombre, carece de significado decir que la media de la variable sexo en un determinado set de datos es $0.5$. En situaciones como esta, no se pueden emplear medidas de similitud basadas en distancia euclídea, manhattan, correlación...
Dadas dos observaciones A y B, cada uno con $n$ atributos binarios, el simple matching coefficient (SMC) define la similitud entre ellos como:
$$SMC= \frac{\text{número coincidencias}}{\text{número total de atributos}} = \frac{M_{00} + M{11}}{M_{00} + M_{01}+ M_{10}+M_{11}}$$donde $M_{00}$ y $M_{11}$ son el número de variables para las que ambas observaciones tienen el mismo valor (ambas $0$ o ambas $1$), y $M_{01}$ y $M_{10}$ el número de variables que no coinciden. El valor de distancia simple matching distance (SMD) se corresponde con $1 - SMC$.
Índice Jaccard¶
El índice Jaccard o coeficiente de correlación Jaccard es similar al simple matching coefficient (SMC). La diferencia radica en que, el SMC, tiene el término $M_{00}$ en el numerador y denominador, mientras que el índice de Jaccard no. Esto significa que SMC considera como coincidencias tanto si el atributo está presente en ambos sets como si el atributo no está en ninguno de los sets, mientras que Jaccard solo cuenta como coincidencias cuando el atributo está presente en ambos sets.
$$J= \frac{M_{11}}{M_{01}+ M_{10}+M_{11}}$$o en términos matemáticos de sets:
$$J(A,B) = \frac{A \cap B}{A \cup B}$$La distancia de Jaccard ($1-J$) supera a la simple matching distance en aquellas situaciones en las que la coincidencia de ausencia no aporta información. Para ilustrar este hecho, supóngase que se quiere cuantificar la similitud entre dos clientes de un supermercado en base a los artículos comprados. Es de esperar que cada cliente solo adquiera unos pocos artículos de los muchos disponibles, por lo que el número de artículos no comprados por ninguno ($M_{00}$) será muy alto. Como la distancia de Jaccard ignora las coincidencias de tipo $M_{00}$, el grado de similitud dependerá únicamente de las coincidencias entre los artículos comprados.
Distancia coseno¶
El coseno del ángulo que forman dos vectores puede interpretarse como una medida de similitud de sus orientaciones, independientemente de sus magnitudes. Si dos vectores tienen exactamente la misma orientación (el ángulo que forman es 0º) su coseno toma el valor de 1, si son perpendiculares (forman un ángulo de 90º) su coseno es 0 y si tienen orientaciones opuestas (ángulo de 180º) su coseno es de -1.
$$\text{cos}(\alpha) = \frac{\textbf{x} \cdot \textbf{y}}{||\textbf{x}|| \ ||\textbf{y}||}$$Si los vectores se normalizan restandoles la media ($X − \bar{X}$), la medida recibe el nombre de coseno centrado y es equivalente a la correlación de Pearson.
Escalado de las variables¶
La escala en la que se miden las variables y la magnitud de su varianza pueden afectar a los resultados obtenidos por clustering. Si una variable tiene una escala mucho mayor que el resto, determinará en gran medida el valor de distancia/similitud obtenido al comparar las observaciones, dirigiendo así la agrupación final. Escalar y centrar las variables antes de calcular la matriz de distancias para que tengan media 0 y desviación estándar 1, asegura que todas las variables tengan el mismo peso cuando se realice el clustering. Sebastian Raschka hace un análisis muy explicativo de los distintos tipos de escalado y normalización.
La forma más utilizada para escalar y centrar las variables es:
$$\frac{x_i - mean(x)}{sd(x)}$$Aplicando esta transformación, existe una relación entre la distancia euclídea y la correlación de Pearson que hace que los resultados obtenidos por clustering sean equivalentes.
$$d_{euclídea}(p,q\ estandarizados) = \sqrt{2n(1-cor(p,q))}$$Ejemplo: cálculo de distancias¶
El set de datos USArrests contiene el porcentaje de asaltos (Assault), asesinatos (Murder) y secuestros (Rape) por cada 100,000 habitantes para cada uno de los 50 estados de USA (1973). Además, también incluye el porcentaje de la población de cada estado que vive en zonas rurales (UrbanPoP). Empleando estas variables, se pretende calcular una matriz de distancias que permita identificar los estados más similares.
Dos de las librerías de python que implementan las distancias descritas en este documento (junto con otras) son sklearn.metrics.pairwise_distances y scipy.spatial.distance. En concreto, sklearn permite calcular las distancias: ‘cityblock’, ‘cosine’, ‘euclidean’, ‘l1’, ‘l2’, ‘manhattan’, ‘braycurtis’, ‘canberra’, ‘chebyshev’, ‘correlation’, ‘dice’, ‘hamming’, ‘jaccard’, ‘kulsinski’, ‘mahalanobis’, ‘minkowski’, ‘rogerstanimoto’, ‘russellrao’, ‘seuclidean’, ‘sokalmichener’, ‘sokalsneath’, ‘sqeuclidean’ y ‘yule’.
Librerías
Las librerías utilizadas en este ejemplo son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
import statsmodels.api as sm
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
plt.style.use('ggplot')
# Preprocesado y modelado
# ==============================================================================
from sklearn.metrics import pairwise_distances
from sklearn.preprocessing import StandardScaler
# Configuración warnings
# ==============================================================================
import warnings
warnings.filterwarnings('once')
Datos
USArrests = sm.datasets.get_rdataset("USArrests", "datasets")
datos = USArrests.data
datos = datos.rename_axis(None, axis=0)
datos.head(4)
| Murder | Assault | UrbanPop | Rape | |
|---|---|---|---|---|
| Alabama | 13.2 | 236 | 58 | 21.2 |
| Alaska | 10.0 | 263 | 48 | 44.5 |
| Arizona | 8.1 | 294 | 80 | 31.0 |
| Arkansas | 8.8 | 190 | 50 | 19.5 |
Cálculo distancias
# Escalado de las variables
# ==============================================================================
scaler = StandardScaler().set_output(transform="pandas")
datos_scaled = scaler.fit_transform(datos)
datos_scaled.head(4)
| Murder | Assault | UrbanPop | Rape | |
|---|---|---|---|---|
| Alabama | 1.255179 | 0.790787 | -0.526195 | -0.003451 |
| Alaska | 0.513019 | 1.118060 | -1.224067 | 2.509424 |
| Arizona | 0.072361 | 1.493817 | 1.009122 | 1.053466 |
| Arkansas | 0.234708 | 0.233212 | -1.084492 | -0.186794 |
# Cálculo de distancias
# ==============================================================================
print('------------------')
print('Distancia euclídea')
print('------------------')
distancias = pairwise_distances(
X = datos_scaled,
metric ='euclidean'
)
# Se descarta la diagonal superior de la matriz
distancias[np.triu_indices(n=distancias.shape[0])] = np.nan
distancias = pd.DataFrame(
distancias,
columns=datos_scaled.index,
index = datos_scaled.index
)
distancias.iloc[:4,:4]
------------------ Distancia euclídea ------------------
| Alabama | Alaska | Arizona | Arkansas | |
|---|---|---|---|---|
| Alabama | NaN | NaN | NaN | NaN |
| Alaska | 2.731204 | NaN | NaN | NaN |
| Arizona | 2.316805 | 2.728061 | NaN | NaN |
| Arkansas | 1.302905 | 2.854730 | 2.74535 | NaN |
Se reestructura la matriz de distancias para poder ordenar los pares de observaciones por orden de distancia.
# Top n observaciones más similares
# ==============================================================================
(
distancias.melt(ignore_index=False, var_name="estado_b", value_name='distancia')
.rename_axis("estado_a")
.reset_index()
.dropna() \
.sort_values('distancia')
.head(3)
)
| estado_a | estado_b | distancia | |
|---|---|---|---|
| 728 | New Hampshire | Iowa | 0.207944 |
| 631 | New York | Illinois | 0.353774 |
| 665 | Kansas | Indiana | 0.433124 |
# Estados con mayor y menor distancia
# ==============================================================================
fig, axs = plt.subplots(1,2, figsize=(8, 3.5))
datos.loc[['Vermont', 'Florida']].transpose().rename_axis(None, axis=1).plot(ax= axs[0])
axs[0].set_title('Estados con mayor distancia')
datos.loc[['New Hampshire', 'Iowa']].transpose().rename_axis(None, axis=1).plot(ax= axs[1])
axs[1].set_title('Estados con menor distancia')
plt.tight_layout();

Número óptimo de clusters¶
Determinar el número óptimo de clusters es uno de los pasos más complicados a la hora de aplicar métodos de clustering, sobre todo cuando se trata de partitioning clustering, donde el número se tiene que especificar antes de poder ver los resultados. No existe una forma única de averiguar el número adecuado de clusters. Es un proceso bastante subjetivo que depende en gran medida del algoritmo empleado y de si se dispone de información previa sobre los datos con los que se está trabajando. A pesar de ello, se han desarrollado varias estrategias que ayudan en el proceso.
Método Elbow¶
El método Elbow, también conocido como método del codo, sigue una estrategia comúnmente empleada para encontrar el valor óptimo de un hiperparámetro. La idea es probar un rango de valores del hiperparámetro en cuestión, representar gráficamente los resultados obtenidos con cada uno, e identificar aquel punto de la curva (codo) a partir del cual la mejora deja de ser notable. En los casos de partitioning clustering, como por ejemplo K-means, las observaciones se agrupan de forma que se minimiza la varianza total intra-cluster. El método Elbow calcula la varianza total intra-cluster en función del número de clusters y escoge como óptimo aquel valor a partir del cual añadir más clusters apenas consigue mejoría.
Método average silhouette¶
El método de average silhouette considera como número óptimo de clusters aquel que maximiza la media del silhouette coeficient de todas las observaciones.
El silhouette coeficient $(s_i)$ cuantifica cómo de buena es la asignación que se ha hecho de una observación comparando su similitud con el resto de observaciones de su cluster frente a las de los otros clusters. Su valor puede estar entre -1 y 1, siendo valores próximos a 1 un indicativo de que la observación se ha asignado al cluster correcto.
Para cada observación $i$, el silhouette coeficient $(s_i)$ se obtiene del siguiente modo:
Calcular el promedio de las distancias (llámese $a_i$) entre la observación $i$ y el resto de observaciones que pertenecen al mismo cluster. Cuanto menor sea $a_i$, mejor ha sido la asignación de $i$ a su cluster.
Calcular la distancia promedio entre la observación $i$ y el resto de clusters. Entendiendo por distancia promedio entre $i$ y un determinado cluster $C$ como la media de las distancias entre $i$ y las observaciones del cluster $C$.
Identificar como $b_i$ a la menor de las distancias promedio entre $i$ y el resto de clusters, es decir, la distancia al cluster más próximo (neighbouring cluster).
Calcular el valor de silhouette como:
Se considera como número óptimo de clusters aquel que maximiza la media del silhouette coeficient de todas las observaciones.
Estadístico gap¶
El estadístico gap fue publicado por R.Tibshirani, G.Walther y T. Hastie, autores también del magnífico libro Introduction to Statistical Learning. Este estadístico compara, para diferentes valores de k, la varianza total intra-cluster observada frente al valor esperado acorde a una distribución uniforme de referencia. La estimación del número óptimo de clusters es el valor k con el que se consigue maximizar el estadístico gap, es decir, encuentra el valor de k con el que se consigue una estructura de clusters lo más alejada posible de una distribución uniforme aleatoria. Este método puede aplicarse a cualquier tipo de clustering.
El algoritmo del gap statistic method es el siguiente:
Hacer clustering de los datos para un rango de valores de $k$ ($k=1$, ..., $K=n$) y calcular para cada uno el valor de la varianza total intra-cluster ($W_k$).
Simular $B$ sets de datos de referencia, todos ellos con una distribución aleatoria uniforme. Aplicar clustering a cada uno de los sets con el mismo rango de valores $k$ empleado en los datos originales, y calculando en cada caso la varianza total intra-cluster ($W_{kb}$). Se recomienda emplear valores de $B=500$.
Calcular el estadístico gap para cada valor de $k$ como la desviación de la varianza observada $W_k$ respecto del valor esperado acorde a la distribución de referencia ($W_{kb}$). Calcular también su desviación estándar.
- Identificar el número de clusters óptimo como el menor de los valores $k$ para el que el estadístico gap se aleja menos de una desviación estándar del valor gap del siguiente $k$: $gap(k) \geq gap(k+1) - s_{k+1}$.
Los métodos Elbow, Silhouette y gap no tienen por qué coincidir exactamente en su estimación del número óptimo de clusters, pero tienden a acotar el rango de posibles valores. Por esta razón es recomendable calcular los tres y en función de los resultados decidir.
K-means¶
El algoritmo K-means (MacQueen, 1967) agrupa las observaciones en un número predefinido de K clusters de forma que, la suma de las varianzas internas de los clusters, sea lo menor posible.
Existen varias implementaciones de este algoritmo, la más común de ellas se conoce como Lloyd’s. En la bibliografía es común encontrar los términos inertia, within-cluster sum-of-squares o varianza intra-cluster para referirse a la varianza interna de los clusters.
Algoritmo¶
Considérense $C_1$,..., $C_K$ como los sets formados por los índices de las observaciones de cada uno de los clusters. Por ejemplo, el set $C_1$ contiene los índices de las observaciones agrupadas en el cluster 1. La nomenclatura empleada para indicar que la observación $i$ pertenece al cluster $k$ es: $i \in C_k$. Todos los sets satisfacen dos propiedades:
$C_1 \cup C_2 \cup ... \cup C_K = \{1,...,n\}$. Significa que toda observación pertenece a uno de los K clusters.
$C_k \cap C_{k'} = \emptyset$ para todo $k \neq k'$. Implica que los clusters no solapan, ninguna observación pertenece a más de un cluster a la vez.
Dos de las medidas comúnmente empleadas para cuantificar la varianza interna de un cluster ($W(C_k)$) son:
- La suma de las distancias euclídeas al cuadrado entre cada observación ($x_i$) y el centroide ($\mu$) de su cluster. Esto equivale a la suma de cuadrados internos del cluster y es la métrica que utiliza scikitlearn.
- La suma de las distancias euclídeas al cuadrado entre todos los pares de observaciones que forman el cluster, dividida entre el número de observaciones del cluster.
Minimizar la suma total de varianza interna $\sum^k_{k=1}W(C_k)$ de forma exacta (encontrar el mínimo global) es un proceso muy complejo debido a la inmensa cantidad de formas en las que n observaciones se pueden repartir en K grupos. En lugar de esto, k-means trata de encontrar una solución que, aun no siendo la mejor de entre todas las posibles, sea buena (óptimo local). El algoritmo empleado es:
Especificar el número K de clusters que se quieren crear.
Seleccionar de forma aleatoria k observaciones del set de datos como centroides iniciales.
Asignar cada una de las observaciones al centroide más cercano.
Para cada uno de los K clusters generados en el paso 3, recalcular su centroide.
Repetir los pasos 3 y 4 hasta que las asignaciones no cambien o se alcance el número máximo de iteraciones establecido.
Este algoritmo garantiza que, en cada paso, se reduzca la intra-varianza total de los clusters hasta alcanzar un óptimo local. Debido a que el algoritmo de K-means no evalúa todas las posibles distribuciones de las observaciones sino solo parte de ellas, los resultados obtenidos dependen de la asignación aleatoria inicial (paso 2). Por esta razón, es importante ejecutar el algoritmo varias veces (25-50), cada una con una asignación aleatoria inicial distinta, y seleccionar aquella que haya conseguido una menor varianza total.
Ventajas y desventajas¶
K-means es uno de los métodos de clustering más utilizados. Destaca por la sencillez y velocidad de su algoritmo, sin embargo, presenta una serie de limitaciones que se deben tener en cuenta.
Requiere que se indique de antemano el número de clusters que se van a crear. Esto puede ser complicado si no se dispone de información adicional sobre los datos con los que se trabaja. Se han desarrollado varias estrategias para ayudar a identificar potenciales valores óptimos de K (elbow, shilouette), pero todas ellas son orientativas.
Dificultad para detectar clusters alargados o con formas irregulares.
Las agrupaciones resultantes pueden variar dependiendo de la asignación aleatoria inicial de los centroides. Para minimizar este problema, se recomienda repetir el proceso de clustering entre 25-50 veces y seleccionar como resultado definitivo el que tenga menor suma total de varianza interna. Aun así, solo se puede garantizar la reproducibilidad de los resultados si se emplean semillas.
Presenta problemas de robustez frente a outliers. La única solución es excluirlos o recurrir a otros métodos de clustering más robustos como K-medoids (PAM).
Ejemplo¶
Los siguientes datos simulados contienen observaciones que pertenecen a cuatro grupos distintos. Se pretende aplicar K-means-clustering con el objetivo de identificar las agrupaciones reales.
Librerías
Las librerías utilizadas en este ejemplo son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
from sklearn.datasets import make_blobs
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
plt.style.use('ggplot')
# Preprocesado y modelado
# ==============================================================================
from sklearn.cluster import KMeans
from sklearn.preprocessing import StandardScaler
from sklearn.metrics import silhouette_score
# Configuración warnings
# ==============================================================================
import warnings
warnings.filterwarnings('once')
Datos
# Simulación de datos
# ==============================================================================
X, y = make_blobs(
n_samples = 300,
n_features = 2,
centers = 4,
cluster_std = 0.60,
shuffle = True,
random_state = 0
)
fig, ax = plt.subplots(1, 1, figsize=(6, 3.5))
ax.scatter(
x = X[:, 0],
y = X[:, 1],
c = 'white',
marker = 'o',
edgecolor = 'black',
)
ax.set_title('Datos simulados');

Modelo Kmeans
Con la clase sklearn.cluster.KMeans de Scikit-Learn se pueden entrenar modelos de clustering utilizando el algoritmo k-means. Entre sus parámetros destacan:
n_clusters: determina el número $K$ de clusters que se van a generar.init: estrategia para asignar los centroides iniciales. Por defecto se emplea 'k-means++', una estrategia que trata de alejar los centroides lo máximo posible facilitando la convergencia. Sin embargo, esta estrategia puede ralentizar el proceso cuando hay muchos datos, si esto ocurre, es mejor utilizar 'random'.n_init: determina el número de veces que se va a repetir el proceso, cada vez con una asignación aleatoria inicial distinta. Es recomendable que este último valor sea alto, entre 10-25, para no obtener resultados subóptimos debido a una iniciación poco afortunada del proceso.max_iter: número máximo de iteraciones permitidas.random_state: semilla para garantizar la reproducibilidad de los resultados.
# Modelo
# ==============================================================================
scaler = StandardScaler().set_output(transform="pandas")
X_scaled = scaler.fit_transform(X)
modelo_kmeans = KMeans(n_clusters=4, n_init=25, random_state=123)
modelo_kmeans.fit(X=X_scaled)
KMeans(n_clusters=4, n_init=25, random_state=123)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| n_clusters | 4 | |
| init | 'k-means++' | |
| n_init | 25 | |
| max_iter | 300 | |
| tol | 0.0001 | |
| verbose | 0 | |
| random_state | 123 | |
| copy_x | True | |
| algorithm | 'lloyd' |
El objeto devuelto por KMeans() contiene entre otros datos: la media de cada una de las variables para cada cluster (cluster_centers_), es decir, los centroides. Un vector indicando a qué cluster se ha asignado cada observación (.labels_) y la suma total de cuadrados internos de todos los clusters (.inertia_).
Número de clusters
Al tratarse de una simulación, se conoce el verdadero número de grupos (4) y a cuál de ellos pertenece cada observación. Esto no sucede en la mayoría de casos prácticos, pero es útil como ejemplo ilustrativo cómo funciona K-means.
# Clasificación con el modelo kmeans
# ==============================================================================
y_predict = modelo_kmeans.predict(X=X_scaled)
# Representación gráfica: grupos originales vs clusters creados
# ==============================================================================
fig, ax = plt.subplots(1, 2, figsize=(9, 4), sharey=True)
# Grupos originales
for i in np.unique(y):
ax[0].scatter(
x = X_scaled.iloc[y == i, 0],
y = X_scaled.iloc[y == i, 1],
c = plt.rcParams['axes.prop_cycle'].by_key()['color'][i],
marker = 'o',
edgecolor = 'black',
label = f"Grupo {i}"
)
ax[0].set_title('Clusters generados por Kmeans')
ax[0].legend()
for i in np.unique(y_predict):
ax[1].scatter(
x = X_scaled.iloc[y_predict == i, 0],
y = X_scaled.iloc[y_predict == i, 1],
c = plt.rcParams['axes.prop_cycle'].by_key()['color'][i],
marker = 'o',
edgecolor = 'black',
label = f"Cluster {i}"
)
ax[1].scatter(
x = modelo_kmeans.cluster_centers_[:, 0],
y = modelo_kmeans.cluster_centers_[:, 1],
c = 'black',
s = 200,
marker = '*',
label = 'centroides'
)
ax[1].set_title('Clusters generados por Kmeans')
ax[1].legend()
plt.tight_layout();

Este tipo de visualización es muy útil e informativa, sin embargo, solo es posible cuando se trabaja con dos dimensiones. Si los datos contienen más de dos variables (dimensiones), una posible solución es utilizar las dos primeras componentes principales obtenidas con un PCA previo.
El número de aciertos y errores puede representarse en modo de matriz de confusión. A la hora de interpretar estas matrices, es importante recordar que el clustering asigna las observaciones a clusters cuyo identificador no tiene porqué coincidir con la nomenclatura empleada para los grupos reales. En este ejemplo, el grupo 1 se ha asignado al cluster 3. Así pues, por cada fila de la matriz cabe esperar un valor alto (coincidencias) para una de las posiciones y valores bajos en las otras (errores de clasificación), pero no tienen por qué coincidir los nombres (diagonal).
# Matriz de confusión: grupos originales vs clusters creados
# ==============================================================================
pd.crosstab(y, y_predict, dropna=False, rownames=['grupo_real'], colnames=['cluster'])
| cluster | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| grupo_real | ||||
| 0 | 75 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 75 |
| 2 | 0 | 75 | 0 | 0 |
| 3 | 0 | 0 | 75 | 0 |
En este análisis, todas las observaciones se han clasificado correctamente. De nuevo repetir que, en la realidad, no se suelen conocer los verdaderos grupos en los que se dividen las observaciones, de lo contrario no se necesitaría aplicar clustering.
Supóngase ahora que se trata de un caso real, en el que se desconoce el número de grupos en los que se subdividen las observaciones. El analista tendría que probar con diferentes valores de $K$ y decidir cuál parece más razonable. A continuación, se muestran los resultados para $K=2$ y $K=6$.
# Resultados para K = 2
# ==============================================================================
fig, ax = plt.subplots(1, 2, figsize=(9, 4), sharey=True)
y_predict = KMeans(n_clusters=2, n_init=25, random_state=123).fit_predict(X=X_scaled)
ax[0].scatter(
x = X_scaled.iloc[:, 0],
y = X_scaled.iloc[:, 1],
c = y_predict,
marker = 'o',
edgecolor = 'black'
)
ax[0].set_title('KMeans K=2');
# Resultados para K = 6
# ==============================================================================
y_predict = KMeans(n_clusters=6, n_init=25, random_state=123).fit_predict(X=X_scaled)
ax[1].scatter(
x = X_scaled.iloc[:, 0],
y = X_scaled.iloc[:, 1],
c = y_predict,
marker = 'o',
edgecolor = 'black'
)
ax[1].set_title('KMeans K=6')
plt.tight_layout();

Al observar los resultados obtenidos para K = 2, es intuitivo pensar que el grupo que se encuentra entorno a las coordenadas $x = 0.5, y = 0$ (mayoritariamente considerado como amarillo) debería ser un grupo separado. Para K = 6 no parece muy razonable la separación de los grupos amarillo y azul. Este ejemplo muestra la principal limitación del método de K-means, el hecho de tener que escoger de antemano el número de clusters que se generan.
La deducción anterior solo puede hacerse visualmente cuando se trata de dos dimensiones. Una forma sencilla de estimar el número K óptimo de clusters cuando no se dispone de esta información, es aplicar el algoritmo de K-means para un rango de valores de K e identificar aquel valor a partir del cual la reducción en la suma total de varianza intra-cluster (inertia) deja de ser sustancial. A esta estrategia se la conoce como método del codo o elbow method.
# Método elbow para identificar el número óptimo de clusters
# ==============================================================================
range_n_clusters = range(1, 15)
inertias = []
for n_clusters in range_n_clusters:
modelo_kmeans = KMeans(
n_clusters = n_clusters,
n_init = 20,
random_state = 123
)
modelo_kmeans.fit(X_scaled)
inertias.append(modelo_kmeans.inertia_)
fig, ax = plt.subplots(1, 1, figsize=(6, 3.5))
ax.plot(range_n_clusters, inertias, marker='o')
ax.set_title("Evolución de la varianza intra-cluster total")
ax.set_xlabel('Número clusters')
ax.set_ylabel('Intra-cluster (inertia)');

A partir de 4 clusters la reducción en la suma total de cuadrados internos parece estabilizarse, indicando que K = 4 es una buena opción.
# Método silhouette para identificar el número óptimo de clusters
# ==============================================================================
range_n_clusters = range(2, 15)
valores_medios_silhouette = []
for n_clusters in range_n_clusters:
modelo_kmeans = KMeans(
n_clusters = n_clusters,
n_init = 20,
random_state = 123
)
cluster_labels = modelo_kmeans.fit_predict(X_scaled)
silhouette_avg = silhouette_score(X_scaled, cluster_labels)
valores_medios_silhouette.append(silhouette_avg)
fig, ax = plt.subplots(1, 1, figsize=(6, 3.5))
ax.plot(range_n_clusters, valores_medios_silhouette, marker='o')
ax.set_title("Evolución de media de los índices silhouette")
ax.set_xlabel('Número clusters')
ax.set_ylabel('Media índices silhouette');

El valor medio de los índices silhouette se maximiza con 4 clusters. Acorde a este criterio, K = 4 es la mejor opción.
Ambos criterios, elbow y silhouette, identifican el valor K=4 como valor óptimo de clusters.
K-medoids¶
Algoritmo¶
K-medoids es un método de clustering muy similar a K-means en cuanto a que ambos agrupan las observaciones en K clusters, donde K es un valor preestablecido por el analista. La diferencia es que, en K-medoids, cada cluster está representado por una observación presente en el cluster (medoid), mientras que en K-means cada cluster está representado por su centroide, que se corresponde con el promedio de todas las observaciones del cluster pero con ninguna en particular.
Una definición más exacta del término medoid es: elemento dentro de un cluster cuya distancia (diferencia) promedio entre él y todos los demás elementos del mismo cluster es lo menor posible. Se corresponde con el elemento más central del cluster y por lo tanto puede considerarse como el más representativo. El hecho de utilizar medoids en lugar de centroides hace de K-medoids un método más robusto que K-means, viéndose menos afectado por outliers o ruido. A modo de idea intuitiva puede considerarse como la analogía entre media y mediana.
El algoritmo más empleado para aplicar K-medoids se conoce como PAM (Partitioning Around Medoids) y sigue los siguientes pasos:
Seleccionar K observaciones aleatorias como medoids iniciales. También es posible identificarlas de forma específica.
Calcular la matriz de distancia entre todas las observaciones si esta no se ha calculado anteriormente.
Asignar cada observación a su medoid más cercano.
Para cada uno de los clusters creados, comprobar si seleccionando otra observación como medoid se consigue reducir la distancia promedio del cluster, si esto ocurre, seleccionar la observación que consigue una mayor reducción como nuevo medoid.
Si al menos un medoid ha cambiado en el paso 4, volver al paso 3, de lo contrario, se termina el proceso.
A diferencia del algoritmo K-means, en el que se minimiza la suma total de cuadrados intra-cluster (suma de las distancias al cuadrado de cada observación respecto a su centroide), el algoritmo K-medoids minimiza la suma de las diferencias de cada observación respecto a su medoid.
Por lo general, el método de K-medoids se utiliza cuando se conoce o se sospecha de la presencia de outliers. Si esto ocurre, es recomendable utilizar como medida de similitud la distancia de Manhattan, ya que es menos sensible a outliers que la euclídea.
Ventajas y desventajas¶
K-medoids es un método de clustering más robusto que K-means, por lo es más adecuado cuando el set de datos contiene outliers o ruido.
Al igual que K-means, necesita que se especifique de antemano el número de clusters que se van a crear. Esto puede ser complicado de determinar si no se dispone de información adicional sobre los datos. Muchas de las estrategias empleadas en K-means para identificar el número óptimo, pueden aplicarse en K-medoids.
Para sets de datos grandes se necesitan muchos recursos computacionales. En tal situación se recomienda aplicar el método CLARA.
El algoritmo K-medoids está implementado en la clase KMedoids de la librería sklearn_extra. Su funcionamiento es análogo al descrito en el apartado de K-means. También está disponible en la librería PyClustering.
Hierarchical clustering¶
Hierarchical clustering es una alternativa a los métodos de partitioning clustering que no requiere que se pre-especifique el número de clusters. Los métodos que engloba el hierarchical clustering se subdividen en dos tipos dependiendo de la estrategia seguida para crear los grupos:
Aglomerativo (agglomerative clustering o bottom-up): el agrupamiento se inicia con todas las observaciones separadas, cada una formando un cluster individual. Los clusters se van combinado a medida que la estructura crece hasta converger en uno solo.
Divisivo (divisive clustering o top-down): es la estrategia opuesta al aglomerativo. Se inicia con todas las observaciones contenidas en un mismo cluster y se suceden divisiones hasta que cada observación forma un cluster* individual.
En ambos casos, los resultados pueden representarse de forma muy intuitiva en una estructura de árbol llamada dendrograma.
Aglomerativo¶
El algoritmo seguido para por el clustering aglomerativo es:
Considerar cada una de las n observaciones como un cluster individual, formando así la base del dendrograma (hojas).
Proceso iterativo hasta que todas las observaciones pertenecen a un único cluster:
2.1 Calcular la distancia entre cada posible par de los n clusters. El investigador debe determinar el tipo de medida empleada para cuantificar la similitud entre observaciones o grupos (distancia y linkage).
2.2 Los dos clusters más similares se fusionan, de forma que quedan n-1 clusters.
Cortar la estructura de árbol generada (dendrograma) a una determinada altura para crear los clusters finales.
Para que el proceso de agrupamiento pueda llevarse a cabo tal como indica el algoritmo anterior, es necesario definir cómo se cuantifica la similitud entre dos clusters. Es decir, se tiene que extender el concepto de distancia entre pares de observaciones para que sea aplicable a pares de grupos, cada uno formado por varias observaciones. A este proceso se le conoce como linkage. A continuación, se describen los 5 tipos de linkage más empleados y sus definiciones.
Complete or Maximum: se calcula la distancia entre todos los posibles pares formados por una observación del cluster A y una del cluster B. La mayor de todas ellas se selecciona como la distancia entre los dos clusters. Se trata de la medida más conservadora (maximal intercluster dissimilarity).
Single or Minimum: se calcula la distancia entre todos los posibles pares formados por una observación del cluster A y una del cluster B. La menor de todas ellas se selecciona como la distancia entre los dos clusters. Se trata de la medida menos conservadora (minimal intercluster dissimilarity).
Average: Se calcula la distancia entre todos los posibles pares formados por una observación del cluster A y una del cluster B. El valor promedio de todas ellas se selecciona como la distancia entre los dos clusters (mean intercluster dissimilarity).
Centroid: Se calcula el centroide de cada uno de los clusters y se selecciona la distancia entre ellos como la distancia entre los dos clusters.
Ward: Se trata de un método general. La selección del par de clusters que se combinan en cada paso del agglomerative hierarchical clustering se basa en el valor óptimo de una función objetivo, pudiendo ser esta última cualquier función definida por el analista. El método Ward's minimum variance es un caso particular en el que el objetivo es minimizar la suma total de varianza intra-cluster. En cada paso, se identifican aquellos 2 clusters cuya fusión conlleva menor incremento de la varianza total intra-cluster. Esta es la misma métrica que se minimiza en K-means.
Los métodos de complete, average y Ward's minimum variance suelen ser los preferidos por los analistas debido a que generan dendrogramas más compensados. Sin embargo, no se puede determinar que uno sea mejor que otro, ya que depende del caso de estudio en cuestión. Por ejemplo, en genómica, se emplea con frecuencia el método de centroides. Junto con los resultados de un proceso de hierarchical clustering siempre hay que indicar qué distancia se ha empleado, así como el tipo de linkage, ya que, dependiendo de estos, los resultados pueden variar en gran medida.
Divisivo¶
El algoritmo más conocido de divisive hierarchical clustering es DIANA (DIvisive ANAlysis Clustering). Este algoritmo se inicia con un único cluster que contiene todas las observaciones. A continuación, se van sucediendo divisiones hasta que cada observación forma un cluster independiente. En cada iteración, se selecciona el cluster con mayor diámetro, entendiendo por diámetro de un cluster la mayor de las diferencias entre dos de sus observaciones. Una vez seleccionado el cluster, se identifica la observación más dispar, que es aquella con mayor distancia promedio respecto al resto de observaciones que forman el cluster. Esta observación inicia el nuevo cluster. Se reasignan las observaciones en función de si están más próximas al nuevo cluster o al resto de la partición, dividiendo así el cluster seleccionado en dos nuevos clusters.
Todas las n observaciones forman un único cluster.
Repetir hasta que haya n clusters:
2.1 Calcular para cada cluster la mayor de las distancias entre pares de observaciones (diámetro del cluster).
2.2 Seleccionar el cluster con mayor diámetro.
2.3 Calcular la distancia media de cada observación respecto a las demás.
2.4 La observación más distante inicia un nuevo cluster.
2.5 Se reasignan las observaciones restantes al nuevo cluster o al viejo dependiendo de cuál está más próximo.
A diferencia del clustering aglomerativo, en el que hay que elegir un tipo de distancia y un método de linkage, en el clustering divisivo solo hay que elegir la distancia, no hay linkage.
Dendrograma¶
Los resultados del hierarchical clustering pueden representarse como un árbol en el que las ramas representan la jerarquía con la que se van sucediendo las uniones de clusters.
Supóngase que se dispone de 45 observaciones en un espacio de dos dimensiones, a los que se les aplica hierarchical clustering para intentar identificar grupos. El siguiente dendrograma representa los resultados obtenidos.
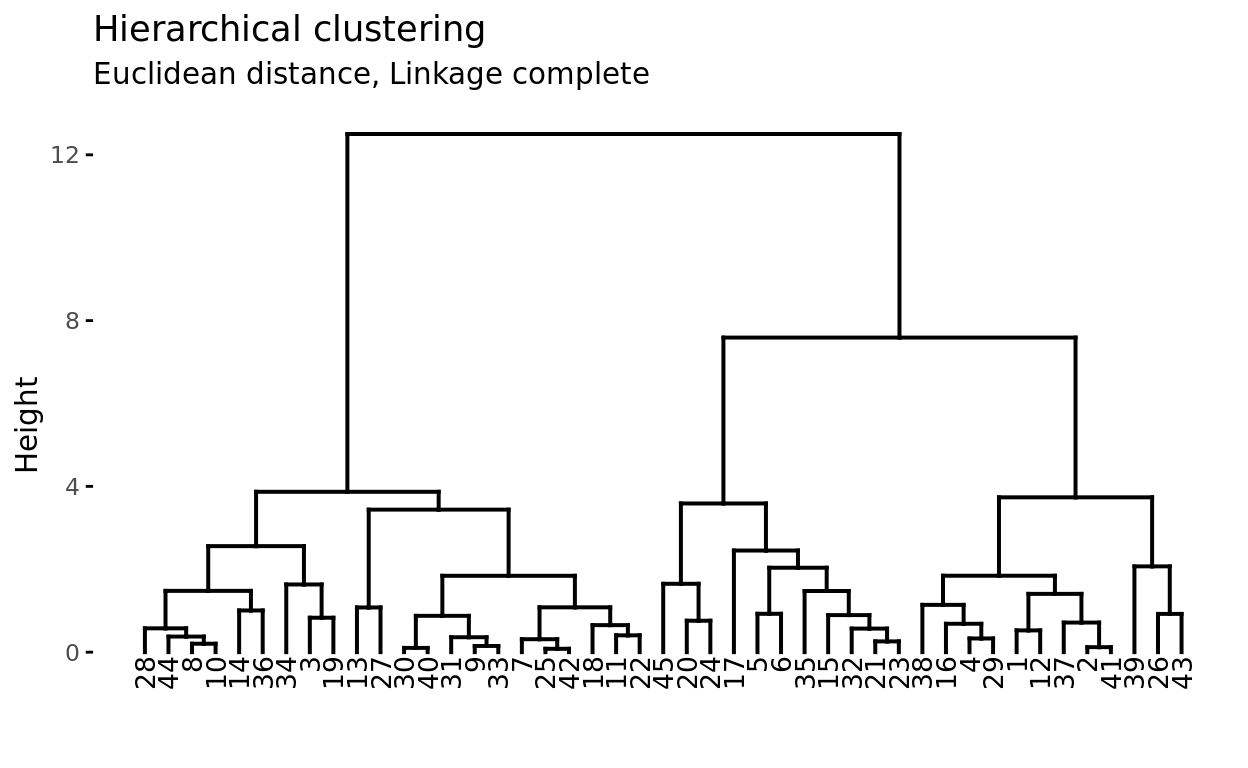
En la base del dendrograma, cada observación forma una terminación individual conocida como hoja o leaf del árbol. A medida que se asciende por la estructura, pares de hojas se fusionan formando las primeras ramas. Estas uniones se corresponden con los pares de observaciones más similares. También ocurre que las ramas se fusionan con otras ramas o con hojas. Cuanto más temprana (más próxima a la base del dendrograma) ocurre una fusión, mayor es la similitud.
Para cualquier par de observaciones, se puede identificar el punto del árbol en el que las ramas que contienen dichas observaciones se fusionan. La altura a la que esto ocurre (eje vertical) indica cómo de similares/diferentes son las dos observaciones. Los dendrogramas, por lo tanto, se deben interpretar únicamente en base al eje vertical y no por las posiciones que ocupan las observaciones en el eje horizontal, esto último es simplemente por estética y puede variar de un programa a otro.
Por ejemplo, la observación 8 es la más similar a la 10 ya que es la primera fusión que recibe la observación 10 (y viceversa). Podría resultar tentador decir que la observación 14, situada inmediatamente a la derecha de la 10, es la siguiente más similar, sin embargo, las observaciones 28 y 44 son más similares a la 10 a pesar de que se encuentran más alejadas en el eje horizontal. Del mismo modo, no es correcto decir que la observación 14 es más similar a la observación 10 de lo que lo es la 36 por el hecho de que está más próxima en el eje horizontal. Prestando atención a la altura en que las respectivas ramas se unen, la única conclusión válida es que la similitud entre los pares 10-14 y 10-36 es la misma.
Cortar el dendograma para generar los clusters¶
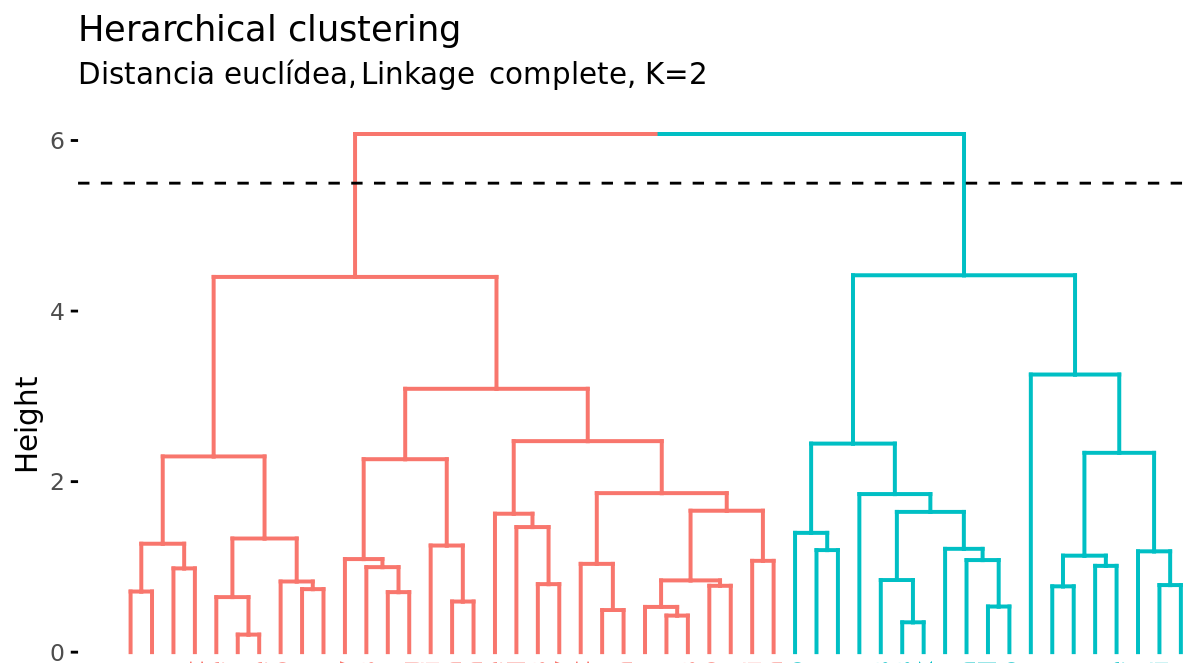
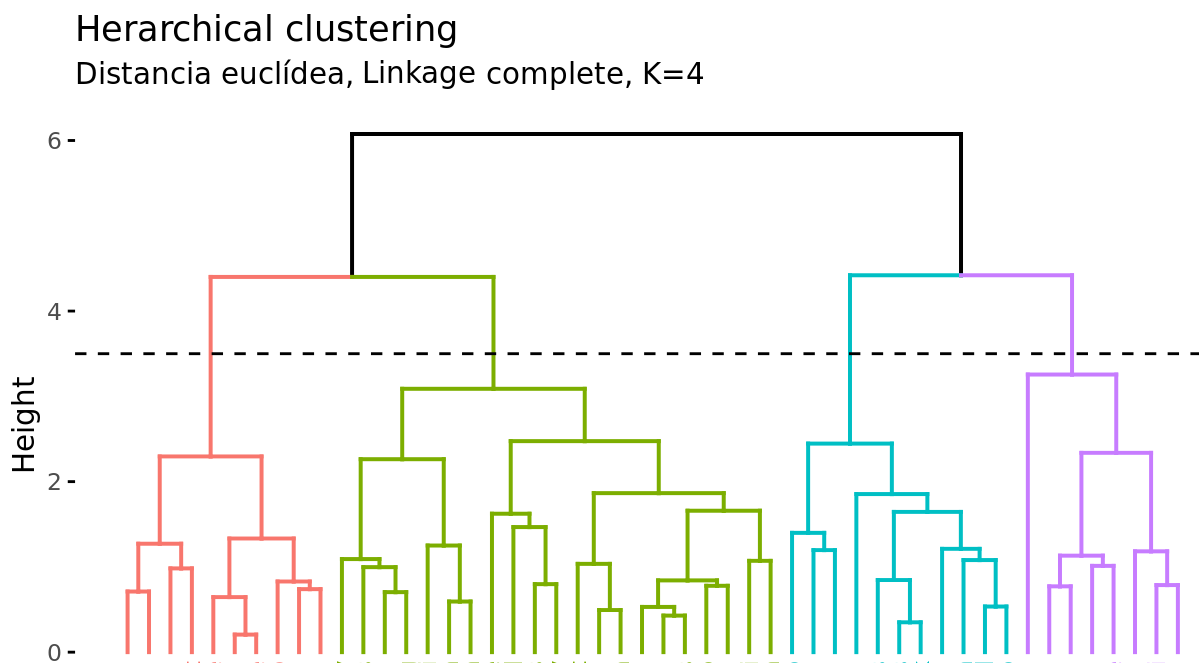
Además de representar en un dendrograma la similitud entre observaciones, se tiene que identificar el número de clusters creados y qué observaciones forman parte de cada uno. Si se realiza un corte horizontal a una determinada altura del dendrograma, el número de ramas que sobrepasan (en sentido ascendente) dicho corte se corresponde con el número de clusters. La siguiente imagen muestra dos veces el mismo dendrograma. Si se realiza el corte a la altura de 5, se obtienen dos clusters, mientras que si se hace a la de 3.5 se obtienen 4. La altura de corte tiene por lo tanto la misma función que el valor K en K-means-clustering: controla el número de clusters obtenidos.
Dos propiedades adicionales se derivan de la forma en que se generan los clusters en el método de hierarchical clustering:
Dada la longitud variable de las ramas, siempre existe un intervalo de altura para el que cualquier corte da lugar al mismo número de clusters. En el ejemplo anterior, todos los cortes entre las alturas 5 y 6 tienen como resultado los mismos 2 clusters.
Con un solo dendrograma se dispone de la flexibilidad para generar cualquier número de clusters desde 1 a n. La selección del número óptimo puede valorarse de forma visual, tratando de identificar las ramas principales en base a la altura a la que ocurren las uniones. En el ejemplo expuesto es razonable elegir entre 2 o 4 clusters.
Ejemplo¶
Los siguientes datos simulados contienen observaciones que pertenecen a cuatro grupos distintos. Se pretende aplicar hierarchical clustering aglomerativo con el objetivo de identificar las agrupaciones reales.
Librerías
Las librerías utilizadas en este ejemplo son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
from sklearn.datasets import make_blobs
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
plt.style.use('ggplot')
# Preprocesado y modelado
# ==============================================================================
from sklearn.cluster import AgglomerativeClustering
from scipy.cluster.hierarchy import dendrogram
from sklearn.preprocessing import StandardScaler
from sklearn.metrics import silhouette_score
# Configuración warnings
# ==============================================================================
import warnings
warnings.filterwarnings('once')
def plot_dendrogram(model, **kwargs):
'''
Esta función extrae la información de un modelo AgglomerativeClustering
y representa su dendograma con la función dendogram de scipy.cluster.hierarchy
'''
counts = np.zeros(model.children_.shape[0])
n_samples = len(model.labels_)
for i, merge in enumerate(model.children_):
current_count = 0
for child_idx in merge:
if child_idx < n_samples:
current_count += 1 # leaf node
else:
current_count += counts[child_idx - n_samples]
counts[i] = current_count
linkage_matrix = np.column_stack([model.children_, model.distances_,
counts]).astype(float)
dendrogram(linkage_matrix, **kwargs)
Datos
# Simulación de datos
# ==============================================================================
X, y = make_blobs(
n_samples = 200,
n_features = 2,
centers = 4,
cluster_std = 0.60,
shuffle = True,
random_state = 0
)
fig, ax = plt.subplots(1, 1, figsize=(6, 3.5))
for i in np.unique(y):
ax.scatter(
x = X[y == i, 0],
y = X[y == i, 1],
c = plt.rcParams['axes.prop_cycle'].by_key()['color'][i],
marker = 'o',
edgecolor = 'black',
label = f"Grupo {i}"
)
ax.set_title('Datos simulados')
ax.legend();

Modelo
Con la clase sklearn.cluster.AgglomerativeClustering de Scikit-Learn se pueden entrenar modelos de clustering utilizando el algoritmo hierarchical clustering aglomerativo. Entre sus parámetros destacan:
n_clusters: determina el número de clusters que se van a generar. En su lugar, su valor puede serNonesi se quiere utilizar el criteriodistance_thresholdpara crear los clusters o crecer todo el dendograma.distance_threshold: distancia (altura del dendograma) a partir de la cual se dejan de unir los clusters. Indicardistance_threshold=0para crecer todo el árbol.compute_full_tree: si se calcula la jerarquía completa de clusters. Debe serTruesidistance_thresholdes distinto deNone.metric: métrica utilizada como distancia. Puede ser: “euclidean”, “l1”, “l2”, “manhattan”, “cosine”, or “precomputed”. Si se utilizalinkage=“ward”, solo se permite “euclidean”.linkage: tipo de linkage utilizado. Puede ser “ward”, “complete”, “average” o “single”.
Al aplicar un hierarchical clustering aglomerativo se tiene que escoger una medida de distancia y un tipo de linkage. A continuación, se comparan los resultados con los linkages complete, ward y average, utilizando la distancia euclídea como métrica de similitud.
# Escalado de datos
# ==============================================================================
scaler = StandardScaler().set_output(transform="pandas")
X_scaled = scaler.fit_transform(X)
# Modelos
# ==============================================================================
modelo_hclust_complete = AgglomerativeClustering(
metric = 'euclidean',
linkage = 'complete',
distance_threshold = 0,
n_clusters = None
)
modelo_hclust_complete.fit(X=X_scaled)
modelo_hclust_average = AgglomerativeClustering(
metric = 'euclidean',
linkage = 'average',
distance_threshold = 0,
n_clusters = None
)
modelo_hclust_average.fit(X=X_scaled)
modelo_hclust_ward = AgglomerativeClustering(
metric = 'euclidean',
linkage = 'ward',
distance_threshold = 0,
n_clusters = None
)
modelo_hclust_ward.fit(X=X_scaled)
AgglomerativeClustering(distance_threshold=0, n_clusters=None)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| n_clusters | None | |
| metric | 'euclidean' | |
| memory | None | |
| connectivity | None | |
| compute_full_tree | 'auto' | |
| linkage | 'ward' | |
| distance_threshold | 0 | |
| compute_distances | False |
# Dendrogramas
# ==============================================================================
fig, axs = plt.subplots(3, 1, figsize=(8, 8))
plot_dendrogram(modelo_hclust_average, color_threshold=0, ax=axs[0])
axs[0].set_title("Distancia euclídea, Linkage average")
plot_dendrogram(modelo_hclust_complete, color_threshold=0, ax=axs[1])
axs[1].set_title("Distancia euclídea, Linkage complete")
plot_dendrogram(modelo_hclust_ward, color_threshold=0, ax=axs[2])
axs[2].set_title("Distancia euclídea, Linkage ward")
plt.tight_layout();

En este caso, los tres tipos de linkage identifican claramente 4 clusters, si bien esto no significa que en los 3 dendrogramas los clusters estén formados por exactamente las mismas observaciones.
Número de clusters
Una forma de identificar el número de clusters, es inspeccionar visualmente el dendograma y decidir a qué altura se corta para generar los clusters. Por ejemplo, para los resultados generados mediante distancia euclídea y linkage ward, parece sensato cortar el dendograma a una altura de entre 5 y 10, de forma que se creen 4 clusters.
fig, ax = plt.subplots(1, 1, figsize=(8, 4))
altura_corte = 6
plot_dendrogram(modelo_hclust_ward, color_threshold=altura_corte, ax=ax)
ax.set_title("Distancia euclídea, Linkage ward")
ax.axhline(y=altura_corte, c = 'black', linestyle='--', label='altura corte')
ax.legend();

Otra forma de identificar potenciales valores óptimos para el número de clusters en modelos hierarchical clustering es mediante los índices silhouette.
# Método silhouette para identificar el número óptimo de clusters
# ==============================================================================
range_n_clusters = range(2, 15)
valores_medios_silhouette = []
for n_clusters in range_n_clusters:
modelo = AgglomerativeClustering(
metric = 'euclidean',
linkage = 'ward',
n_clusters = n_clusters
)
cluster_labels = modelo.fit_predict(X_scaled)
silhouette_avg = silhouette_score(X_scaled, cluster_labels)
valores_medios_silhouette.append(silhouette_avg)
fig, ax = plt.subplots(1, 1, figsize=(6, 3.5))
ax.plot(range_n_clusters, valores_medios_silhouette, marker='o')
ax.set_title("Evolución de media de los índices silhouette")
ax.set_xlabel('Número clusters')
ax.set_ylabel('Media índices silhouette');

Una vez identificado el número óptimo de clusters, se reentrena el modelo indicando este valor.
# Modelo
# ==============================================================================
modelo_hclust_ward = AgglomerativeClustering(
metric = 'euclidean',
linkage = 'ward',
n_clusters = 4
)
modelo_hclust_ward.fit(X=X_scaled)
AgglomerativeClustering(n_clusters=4)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| n_clusters | 4 | |
| metric | 'euclidean' | |
| memory | None | |
| connectivity | None | |
| compute_full_tree | 'auto' | |
| linkage | 'ward' | |
| distance_threshold | None | |
| compute_distances | False |
Density based clustering (DBSCAN)¶
Density-based spatial clustering of applications with noise (DBSCAN) fue presentado en 1996 por Ester et al. como una forma de identificar clusters siguiendo el modo intuitivo en el que lo hace el cerebro humano, identificando regiones con alta densidad de observaciones separadas por regiones de baja densidad.
Véase la siguiente representación bidimensional de los datos multishape del paquete factoextra de R.
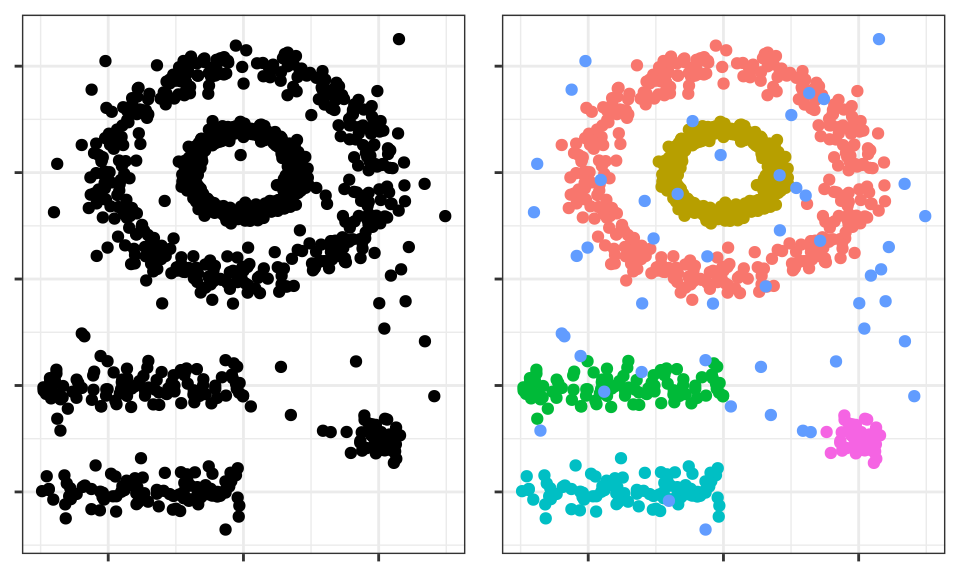
El cerebro humano identifica fácilmente 5 agrupaciones y algunas observaciones aisladas (ruido). Véanse ahora los clusters que se obtienen si se aplica, por ejemplo, K-means clustering.
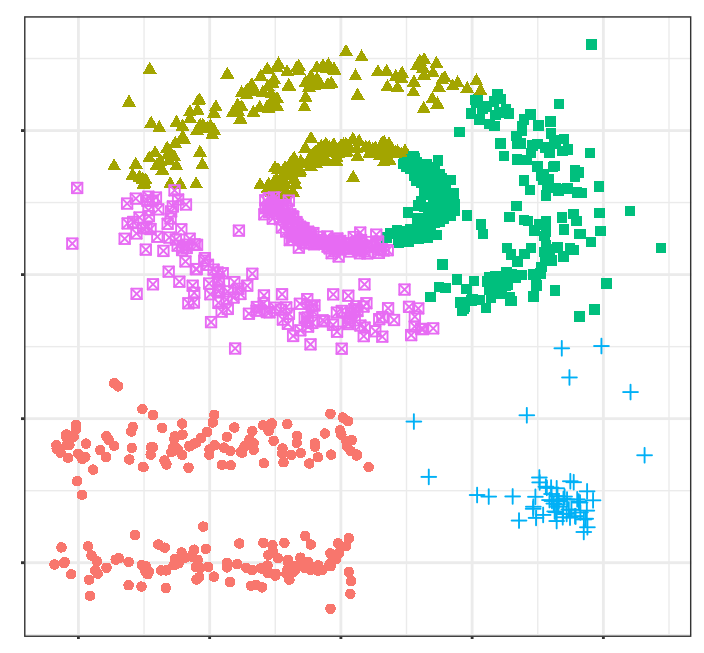
Los clusters generados distan mucho de representar las verdaderas agrupaciones. Esto es así porque los métodos de partitioning clustering como k-means, hierarchical, k-medoids, ... son buenos encontrando agrupaciones con forma esférica o convexa que no contengan un exceso de outliers o ruido, pero fallan al tratar de identificar formas arbitrarias. De ahí que el único cluster que se corresponde con un grupo real sea el amarillo.
DBSCAN evita este problema siguiendo la idea de que, para que una observación forme parte de un cluster, tiene que haber un mínimo de observaciones vecinas dentro de un radio de proximidad y de que los clusters están separados por regiones vacías o con pocas observaciones.
El algoritmo DBSCAN necesita dos parámetros:
Epsilon $(\epsilon)$: radio que define la región vecina a una observación, también llamada $\epsilon$-neighborhood.
Minimum points (min_samples): número mínimo de observaciones dentro de la región epsilon.
Empleando estos dos parámetros, cada observación del set de datos se puede clasificar en una de las siguientes tres categorías:
Core point: observación que tiene en su $\epsilon$-neighborhood un número de observaciones vecinas igual o mayor a min_samples.
Border point: observación no satisface el mínimo de observaciones vecinas para ser core point pero que pertenece al $\epsilon$-neighborhood de otra observación que sí es core point.
Noise u outlier: observación que no es core point ni border point.
Por último, empleando las tres categorías anteriores se pueden definir tres niveles de conectividad entre observaciones:
Directamente alcanzable (direct density reachable): una observación $A$ es directamente alcanzable desde otra observación $B$ si $A$ forma parte del $\epsilon$-neighborhood de $B$ y $B$ es un core point. Por definición, las observaciones solo pueden ser directamente alcanzables desde un core point.
Alcanzable (density reachable): una observación $A$ es alcanzable desde otra observación $B$ si existe una secuencia de core points que van desde $B$ a $A$.
Densamente conectadas (density conected): dos observaciones $A$ y $B$ están densamente conectadas si existe una observación core point $C$ tal que $A$ y $B$ son alcanzables desde $C$.
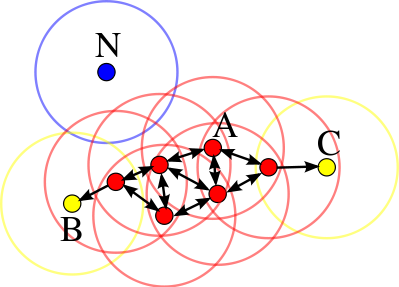
La siguiente imagen muestra las conexiones existentes entre un conjunto de observaciones si se emplea $min_samples = 4$. La observación $A$ y el resto de observaciones marcadas en rojo son core points, ya que todas ellas contienen al menos 4 observaciones vecinas (incluyéndose a ellas mismas) en su $\epsilon$-neighborhood. Como todas son alcanzables entre ellas, forman un cluster. Las observaciones $B$ y $C$ no son core points pero son alcanzables desde $A$ a través de otros core points, por lo tanto, pertenecen al mismo cluster que $A$. La observación $N$ no es ni un core point ni es directamente alcanzable, por lo que se considera como ruido.
Algoritmo¶
Para cada observación $x_i$ calcular la distancia entre ella y el resto de observaciones. Si en su $\epsilon$-neighborhood hay un número de observaciones $\geq min_samples$ marcar la observación como core point, de lo contrario marcarla como visitada.
Para cada observación $x_i$ marcada como core point, si todavía no ha sido asignada a ningún cluster, crear uno nuevo y asignarla a él. Encontrar recursivamente todas las observaciones densamente conectadas a ella y asignarlas al mismo cluster.
Iterar el mismo proceso para todas las observaciones que no hayan sido visitadas.
Aquellas observaciones que tras haber sido visitadas no pertenecen a ningún cluster se marcan como outliers.
Como resultado, todo cluster cumple dos propiedades: todos los puntos que forman parte de un mismo cluster están densamente conectados entre ellos y, si una observación $A$ es densamente alcanzable desde cualquier otra observación de un cluster, entonces $A$ también pertenece al cluster.
Hiperparámetros¶
Como ocurre en muchas otras técnicas estadísticas, en DBSCAN no existe una forma única y exacta de encontrar el valor adecuado de epsilon $(\epsilon)$ y $min_samples$. A modo orientativo se pueden seguir las siguientes premisas:
$min_samples$: cuanto mayor sea el tamaño del set de datos, mayor debe ser el valor mínimo de observaciones vecinas. En el libro Practical Guide to Cluster Analysis in R recomiendan no bajar nunca de 3. Si los datos contienen un nivel alto de ruido, aumentar $min_samples$ favorecerá la creación de clusters significativos menos influenciados por outliers.
epsilon: una buena forma de escoger el valor de $\epsilon$ es estudiar las distancias promedio entre las $k = min_samples$ observaciones más próximas. Al representar estas distancias en función de $\epsilon$, el punto de inflexión de la curva suele ser un valor óptimo. Si el valor de $\epsilon$ escogido es muy pequeño, una proporción alta de las observaciones no se asignarán a ningún cluster, por el contrario, si el valor es demasiado grande, la mayoría de observaciones se agruparán en un único cluster.
Ventajas y desventajas¶
Ventajas
No requiere que el usuario especifique el número de clusters.
Es independiente de la forma que tengan los clusters.
Puede identificar outliers, por lo que los clusters generados no se ven influenciados por ellos.
Desventajas
Es un método determinístico siempre y cuando el orden de los datos sea el mismo. Los border points que son alcanzables desde más de un cluster pueden asignarse a uno u otro dependiendo del orden en el que se procesen los datos.
No genera buenos resultados cuando la densidad de los grupos es muy distinta, ya que no es posible encontrar los parámetros $\epsilon$ y min_samples que sirvan para todos a la vez.
Ejemplo¶
El set de datos multishape del paquete de R factoextra contiene observaciones que pertenecen a 5 grupos distintos junto con cierto ruido (outliers). Como se espera que la distribución espacial de los grupos no sea esférica, se aplica el método de clustering DBSCAN.
Librerías
Las librerías utilizadas en este ejemplo son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
from sklearn.datasets import make_blobs
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
plt.style.use('ggplot')
# Preprocesado y modelado
# ==============================================================================
from sklearn.cluster import DBSCAN
from sklearn.preprocessing import StandardScaler
from sklearn.metrics import silhouette_score
# Configuración warnings
# ==============================================================================
import warnings
warnings.filterwarnings('once')
Datos
url = 'https://raw.githubusercontent.com/JoaquinAmatRodrigo/' \
+ 'Estadistica-machine-learning-python/master/data/multishape.csv'
datos = pd.read_csv(url)
datos.head()
| x | y | shape | |
|---|---|---|---|
| 0 | -0.803739 | -0.853053 | 1 |
| 1 | 0.852851 | 0.367618 | 1 |
| 2 | 0.927180 | -0.274902 | 1 |
| 3 | -0.752626 | -0.511565 | 1 |
| 4 | 0.706846 | 0.810679 | 1 |
Modelo
Con la clase sklearn.cluster.DBSCAN de Scikit-Learn se pueden entrenar modelos de clustering utilizando el algoritmo DBSCAN. Entre sus parámetros destacan:
eps: el radio epsilon que define la vecindad de cada observación.min_samples: el número mínimo de observaciones requeridas en el radio epsilon para que un punto sea considerado como core point.metric: métrica utilizada como distancia. Puede ser: "euclidean", "l1", "l2", "manhattan", "cosine", or "precomputed".
# Escalado de datos
# ==============================================================================
X = datos.drop(columns='shape')
scaler = StandardScaler().set_output(transform="pandas")
X_scaled = scaler.fit_transform(X)
# Modelo
# ==============================================================================
modelo_dbscan = DBSCAN(
eps = 0.2,
min_samples = 5,
metric = 'euclidean',
)
modelo_dbscan.fit(X=X_scaled)
DBSCAN(eps=0.2)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| eps | 0.2 | |
| min_samples | 5 | |
| metric | 'euclidean' | |
| metric_params | None | |
| algorithm | 'auto' | |
| leaf_size | 30 | |
| p | None | |
| n_jobs | None |
# Clasificación
# ==============================================================================
labels = modelo_dbscan.labels_
fig, ax = plt.subplots(1, 1, figsize=(4.5, 4.5))
ax.scatter(
x = X.iloc[:, 0],
y = X.iloc[:, 1],
c = labels,
marker = 'o',
edgecolor = 'black'
)
# Los outliers se identifican con el label -1
ax.scatter(
x = X.iloc[labels == -1, 0],
y = X.iloc[labels == -1, 1],
c = 'red',
marker = 'o',
edgecolor = 'black',
label = 'outliers'
)
ax.legend()
ax.set_title('Clusterings generados por DBSCAN');

# Número de clusters y observaciones "outliers"
# ==============================================================================
n_clusters = len(set(labels)) - (1 if -1 in labels else 0)
n_noise = list(labels).count(-1)
print(f'Número de clusters encontrados: {n_clusters}')
print(f'Número de outliers encontrados: {n_noise}')
Número de clusters encontrados: 5 Número de outliers encontrados: 25
Gaussian mixture models (GMMs)¶
Algoritmo¶
Un Gaussian Mixture model es un modelo probabilístico en el que se considera que las observaciones siguen una distribución probabilística formada por la combinación de múltiples distribuciones normales (componentes). En su aplicación al clustering, puede entenderse como una generalización de K-means con la que, en lugar de asignar cada observación a un único cluster, se obtiene una distribución probabilidad de pertenencia a cada uno.
Para estimar los parámetros que definen la función de distribución de cada cluster (media y matriz de covarianza) se recurre al algoritmo de Expectation-Maximization (EM). Una vez aprendidos los parámetros, se puede calcular la probabilidad que tiene cada observación de pertenecer a cada cluster y asignarla a aquel con mayor probabilidad.
Junto con el número de clusters (componentes), hay que determinar el tipo de matriz de covarianza que pueden tener los clusters. Dependiendo del tipo de matriz, la forma de los clusters puede ser:
tied: todos los clusters comparten la misma matriz de covarianza.
diagonal: las dimensiones de cada cluster a lo largo de cada dimensión puede ser distinto, pero las elipses generadas siempre quedan alineadas con los ejes, es decir, su orientaciones son limitadas.
spherical: las dimensiones de cada cluster son las mismas en todas las dimensiones. Esto permite generar clusters de distinto tamaño pero todos esféricos.
full: cada cluster puede puede ser modelado como una elipse cualquier orientación y dimensiones.
# Función auxiliar para dibujar elipses de covarianza
# ==============================================================================
from sklearn.mixture import GaussianMixture
from matplotlib.patches import Ellipse
import matplotlib.pyplot as plt
def draw_covariance_ellipses(gmm, ax, n_std=3, **kwargs):
"""
Dibuja elipses de covarianza para un modelo GMM.
Parameters
----------
gmm : GaussianMixture
Modelo de mezcla gaussiana ajustado
ax : matplotlib.axes.Axes
Eje donde dibujar
n_std : int
Número máximo de desviaciones estándar a dibujar
**kwargs : dict
Argumentos adicionales para Ellipse (color, alpha, etc.)
"""
ax = ax or plt.gca()
colors = plt.cm.Set2(np.linspace(0, 1, n_std))
for n in range(gmm.n_components):
# Seleccionar matriz de covarianza según el tipo
if gmm.covariance_type == 'full':
cov = gmm.covariances_[n]
elif gmm.covariance_type == 'tied':
cov = gmm.covariances_
elif gmm.covariance_type == 'diag':
cov = np.diag(gmm.covariances_[n])
elif gmm.covariance_type == 'spherical':
cov = np.eye(gmm.means_.shape[1]) * gmm.covariances_[n]
# Calcular parámetros de la elipse usando descomposición en valores propios
# (más eficiente y estable numéricamente que SVD para matrices simétricas)
if cov.shape == (2, 2):
eigenvalues, eigenvectors = np.linalg.eigh(cov)
order = eigenvalues.argsort()[::-1]
eigenvalues, eigenvectors = eigenvalues[order], eigenvectors[:, order]
angle = np.degrees(np.arctan2(eigenvectors[1, 0], eigenvectors[0, 0]))
width, height = 2 * np.sqrt(eigenvalues)
else:
angle = 0
width, height = 2 * np.sqrt(cov)
# Dibujar elipses para 1σ, 2σ, 3σ
for i, nsig in enumerate(range(1, n_std + 1)):
ellipse = Ellipse(
xy=gmm.means_[n],
width=nsig * width,
height=nsig * height,
angle=angle,
facecolor=colors[i],
edgecolor='darkblue',
linewidth=1.5,
alpha=0.25,
**kwargs
)
ax.add_patch(ellipse)
# Marcar el centroide
ax.plot(gmm.means_[n, 0], gmm.means_[n, 1], 'ko', markersize=8,
markerfacecolor='red', markeredgewidth=1.5, zorder=5)
# Comparación visual de tipos de covarianza en GMM
# ==============================================================================
fig, axes = plt.subplots(1, 3, figsize=(8, 3.5), sharey=True)
# Generar datos sintéticos con correlación
rng = np.random.RandomState(5)
X_demo = np.dot(rng.randn(500, 2), rng.randn(2, 2))
covariance_types = ['spherical', 'diag', 'full']
for i, cov_type in enumerate(covariance_types):
# Ajustar modelo con un solo componente
model = GaussianMixture(
n_components = 1,
covariance_type = cov_type,
random_state = 42
).fit(X_demo)
# Graficar datos
axes[i].scatter(
X_demo[:, 0],
X_demo[:, 1],
alpha = 0.4,
s = 30,
c = 'steelblue',
edgecolor = 'white',
linewidth = 0.5
)
# Dibujar elipses de covarianza
draw_covariance_ellipses(model, axes[i])
# Configurar ejes
axes[i].set_xlim(-3.5, 3.5)
axes[i].set_ylim(-3.5, 3.5)
axes[i].set_title(f'covariance_type="{cov_type}"', fontsize=10)
axes[i].set_xlabel('Variable 1')
axes[i].grid(True, alpha=0.3)
axes[i].set_aspect('equal')
# Solo mostrar ylabel en el primer subplot
if i == 0:
axes[i].set_ylabel('Variable 2')
fig.suptitle('Comparación de tipos de matriz de covarianza en GMM', fontsize=14)
plt.tight_layout()
plt.show()

Ejemplo¶
Los siguientes datos simulados contienen observaciones que pertenecen a cuatro grupos distintos. Se pretende aplicar clustering basado en un Gaussian mixture model con el objetivo de identificar las agrupaciones reales.
Librerías
Las librerías utilizadas en este ejemplo son:
# Tratamiento de datos
# ==============================================================================
import numpy as np
import pandas as pd
from sklearn.datasets import make_blobs
# Gráficos
# ==============================================================================
import matplotlib.pyplot as plt
import matplotlib as mpl
from matplotlib.patches import Ellipse
from matplotlib import style
# Preprocesado y modelado
# ==============================================================================
from sklearn.mixture import GaussianMixture
from sklearn.preprocessing import StandardScaler
from sklearn.metrics import silhouette_score
# Configuración warnings
# ==============================================================================
import warnings
warnings.filterwarnings('once')
Datos
# Simulación de datos
# ==============================================================================
X, y = make_blobs(
n_samples = 300,
n_features = 2,
centers = 4,
cluster_std = 0.60,
shuffle = True,
random_state = 0
)
fig, ax = plt.subplots(1, 1, figsize=(6, 3.5))
ax.scatter(
x = X[:, 0],
y = X[:, 1],
c = 'white',
marker = 'o',
edgecolor = 'black',
)
ax.set_title('Datos simulados');
Modelo
Con la clase sklearn.mixture.GaussianMixture de Scikit-Learn se pueden entrenar modelos de GMM utilizando el algoritmo Expectation-Maximization (EM). Entre sus parámetros destacan:
n_components: número de componentes (en este caso clusters) que forman el modelo.covariance_type: tipo de matriz de covarianza (‘full’ (default), ‘tied’, ‘diag’, ‘spherical’).max_iter: número máximo de iteraciones permitidas.random_state: semilla para garantizar la reproducibilidad de los resultados.
# Modelo
# ==============================================================================
modelo_gmm = GaussianMixture(n_components=4, covariance_type='full', random_state=123)
modelo_gmm.fit(X=X)
GaussianMixture(n_components=4, random_state=123)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| n_components | 4 | |
| covariance_type | 'full' | |
| tol | 0.001 | |
| reg_covar | 1e-06 | |
| max_iter | 100 | |
| n_init | 1 | |
| init_params | 'kmeans' | |
| weights_init | None | |
| means_init | None | |
| precisions_init | None | |
| random_state | 123 | |
| warm_start | False | |
| verbose | 0 | |
| verbose_interval | 10 |
El objeto devuelto por GaussianMixture contiene entre otros datos: el peso de cada componente (cluster) en el modelo (weights_), su media (means_) y matriz de covarianza (covariances_).La estructura de esta última depende del tipo de matriz empleada en el ajuste del modelo (covariance_type).
# Media de cada componente
# ==============================================================================
modelo_gmm.means_
array([[ 0.93842466, 4.41564635],
[-1.37355181, 7.75436785],
[ 1.98299679, 0.86735608],
[-1.58913964, 2.82465347]])
# Matriz de covarianza de cada componente
# ==============================================================================
modelo_gmm.covariances_
array([[[ 0.3808649 , -0.02231979],
[-0.02231979, 0.34881473]],
[[ 0.41216925, 0.02884065],
[ 0.02884065, 0.37959193]],
[[ 0.33998651, -0.02620931],
[-0.02620931, 0.34588507]],
[[ 0.32360185, 0.01027908],
[ 0.01027908, 0.30862196]]])
Predicción y clasificación
Una vez entrenado el modelo GMM, se puede predecir la probabilidad que tiene cada observación de pertenecer a cada una de las componentes (clusters). A diferencia de K-means (hard clustering), GMM proporciona una clasificación probabilística (soft clustering), donde cada observación tiene una probabilidad de pertenencia a cada cluster. Para obtener la clasificación final (hard assignment), se asigna cada observación a la componente con mayor probabilidad.
# Probabilidades
# ==============================================================================
# Cada fila es una observación y cada columna la probabilidad de pertenecer a
# cada una de las componentes.
probabilidades = modelo_gmm.predict_proba(X)
probabilidades
array([[2.59296402e-02, 8.04672295e-21, 9.71688955e-01, 2.38140471e-03],
[7.15846263e-09, 9.99999993e-01, 7.13116691e-33, 2.34837535e-15],
[9.99999970e-01, 2.04767765e-08, 8.78022033e-12, 9.13480329e-09],
...,
[9.99965893e-01, 8.75462809e-09, 4.92339895e-10, 3.40976171e-05],
[3.01241053e-06, 9.99996988e-01, 6.44915198e-30, 1.52780115e-18],
[4.39280339e-07, 4.05180546e-15, 1.99754828e-11, 9.99999561e-01]],
shape=(300, 4))
# Clasificación (asignación a la componente de mayor probabilidad)
# ==============================================================================
# Cada fila es una observación y cada columna la probabilidad de pertenecer a
# cada una de las componentes.
clasificacion = modelo_gmm.predict(X)
clasificacion
array([2, 1, 0, 1, 2, 2, 3, 0, 1, 1, 3, 1, 0, 1, 2, 0, 0, 2, 3, 3, 2, 2,
0, 3, 3, 0, 2, 0, 3, 0, 1, 1, 0, 1, 1, 1, 1, 1, 3, 2, 0, 3, 0, 0,
3, 3, 1, 3, 1, 2, 3, 2, 1, 2, 2, 3, 1, 3, 1, 2, 1, 0, 1, 3, 3, 3,
1, 2, 1, 3, 0, 3, 1, 3, 3, 1, 3, 0, 2, 1, 2, 0, 2, 2, 1, 0, 2, 0,
1, 1, 0, 2, 1, 3, 3, 0, 2, 2, 0, 3, 1, 2, 1, 2, 0, 2, 2, 0, 1, 0,
3, 3, 2, 1, 2, 0, 1, 2, 2, 0, 3, 2, 3, 2, 2, 2, 2, 3, 2, 3, 1, 3,
3, 2, 1, 3, 3, 1, 0, 1, 1, 3, 0, 3, 0, 3, 1, 0, 1, 1, 1, 0, 1, 0,
2, 3, 1, 3, 2, 0, 1, 0, 0, 2, 0, 3, 3, 0, 2, 0, 0, 1, 2, 0, 3, 1,
2, 2, 0, 3, 2, 0, 3, 3, 0, 0, 0, 0, 2, 1, 0, 3, 0, 0, 3, 3, 3, 0,
3, 1, 0, 3, 2, 3, 0, 1, 3, 1, 0, 1, 0, 3, 0, 0, 1, 3, 3, 2, 2, 0,
1, 2, 2, 3, 2, 3, 0, 1, 1, 0, 0, 1, 0, 2, 3, 0, 2, 3, 1, 3, 2, 0,
2, 1, 1, 1, 1, 3, 3, 1, 0, 3, 2, 0, 3, 3, 3, 2, 2, 1, 0, 0, 3, 2,
1, 3, 0, 1, 0, 2, 2, 3, 3, 0, 2, 2, 2, 0, 1, 1, 2, 2, 0, 2, 2, 2,
1, 3, 1, 0, 2, 2, 1, 1, 1, 2, 2, 0, 1, 3])
# Visualización de los componentes y distribución del modelo GMM
# ==============================================================================
fig, axs = plt.subplots(1, 2, figsize=(9, 4), constrained_layout=True)
# Panel izquierdo: Distribución de probabilidad de cada componente
for i in np.unique(clasificacion):
axs[0].scatter(
X[clasificacion == i, 0],
X[clasificacion == i, 1],
c=plt.rcParams['axes.prop_cycle'].by_key()['color'][i],
marker='o',
edgecolor='black',
label=f"Componente {i}",
s=50,
alpha=0.7
)
draw_covariance_ellipses(modelo_gmm, ax=axs[0], n_std=2)
axs[0].set_title('Distribución de probabilidad de cada componente', fontsize=11)
axs[0].set_xlabel('Variable 1')
axs[0].set_ylabel('Variable 2')
axs[0].legend()
axs[0].grid(True, alpha=0.3)
# Panel derecho: Distribución de probabilidad del modelo completo
xs = np.linspace(X[:, 0].min(), X[:, 0].max(), 100)
ys = np.linspace(X[:, 1].min(), X[:, 1].max(), 100)
xx, yy = np.meshgrid(xs, ys)
# Calcular scores y convertir de log-probabilidad a probabilidad
log_scores = modelo_gmm.score_samples(np.c_[xx.ravel(), yy.ravel()])
scores = np.exp(log_scores)
# Graficar puntos y contornos
axs[1].scatter(
X[:, 0],
X[:, 1],
c=clasificacion,
cmap='tab10',
s=20,
alpha=0.6,
edgecolor='k',
linewidth=0.5
)
contours = axs[1].contour(
xx, yy,
scores.reshape(xx.shape),
levels=np.percentile(scores, np.linspace(10, 90, 8)),
colors='blue',
alpha=0.4,
linewidths=1.5
)
axs[1].clabel(contours, inline=True, fontsize=8, fmt='%.2e')
axs[1].set_title('Distribución de probabilidad del modelo completo', fontsize=11)
axs[1].set_xlabel('Variable 1')
axs[1].set_ylabel('Variable 2')
axs[1].grid(True, alpha=0.3)
plt.show()

Número de clusters
Dado que los modelos GMM son modelos probabilísticos, se puede recurrir a métricas como el Akaike information criterion (AIC) o Bayesian information criterion (BIC) para identificar cómo de bien se ajustan los datos observados a modelo creado.
n_components = range(1, 21)
valores_bic = []
valores_aic = []
for i in n_components:
modelo = GaussianMixture(n_components=i, covariance_type="full")
modelo = modelo.fit(X)
valores_bic.append(modelo.bic(X))
valores_aic.append(modelo.aic(X))
fig, ax = plt.subplots(1, 1, figsize=(6, 3.84))
ax.plot(n_components, valores_bic, label='BIC')
ax.plot(n_components, valores_aic, label='AIC')
ax.set_title("Valores BIC y AIC")
ax.set_xlabel("Número componentes")
ax.legend();

print(f"Número óptimo acorde al BIC: {range(1, 21)[np.argmin(valores_bic)]}")
print(f"Número óptimo acorde al AIC: {range(1, 21)[np.argmin(valores_aic)]}")
Número óptimo acorde al BIC: 4 Número óptimo acorde al AIC: 4
Ambas métricas identifican el 4 como número óptimo de clusters (componentes).
Limitaciones del clustering¶
El clustering puede ser una herramienta muy útil para encontrar agrupaciones en los datos, sobre todo a medida que el volumen de los mismos aumenta. Sin embargo, es importante recordar sus limitaciones o problemas que pueden surgir al aplicarlo. Algunas de ellas son:
Pequeñas decisiones pueden tener grandes consecuencias:a la hora de utilizar los métodos de clustering se tienen que tomar decisiones que influyen en gran medida en los resultados obtenidos. No existe una única respuesta correcta, por lo que en la práctica se prueban diferentes opciones.
Escalado y centrado de las variables
Qué medida de distancia/similitud emplear
Número de clusters
Tipo de linkage empleado en hierarchical clustering
A que altura establecer el corte de un dendrograma
Validación de los clusters obtenidos: no es fácil comprobar la validez de los resultados ya que en la mayoría de escenarios se desconoce la verdadera agrupación.
Falta de robustez: los métodos de K-means-clustering e hierarchical clustering asignan obligatoriamente cada observación a un grupo. Si existe en la muestra algún outlier, a pesar de que realmente no pertenezca a ningún grupo, el algoritmo lo asignará a uno de ellos provocando una distorsión significativa del cluster en cuestión. Algunas alternativas son k-medoids y DBSCAN.
La naturaleza greedy del algoritmo de hierarchical clustering conlleva que, si se realiza una mala fusión (en clustering aglomerativo) o división (en clustering divisivo) en los pasos iniciales, no se pueda corregir en los pasos siguientes, lo que puede llevar a resultados subóptimos.
Información de sesión¶
import session_info
session_info.show(html=False)
----- matplotlib 3.10.8 numpy 2.2.6 pandas 2.3.3 scipy 1.15.3 session_info v1.0.1 sklearn 1.7.2 statsmodels 0.14.6 ----- IPython 9.8.0 jupyter_client 8.7.0 jupyter_core 5.9.1 ----- Python 3.13.11 | packaged by Anaconda, Inc. | (main, Dec 10 2025, 21:28:48) [GCC 14.3.0] Linux-6.14.0-37-generic-x86_64-with-glibc2.39 ----- Session information updated at 2025-12-29 15:48
Bibliografía¶
Introduction to Machine Learning with Python: A Guide for Data Scientists
Python Data Science Handbook by Jake VanderPlas
Linear Models with R by Julian J.Faraway
An Introduction to Statistical Learning: with Applications in R (Springer Texts in Statistics)
Points of Significance Clustering by Naomi Altman & Martin Krzywinski
Instrucciones para citar¶
¿Cómo citar este documento?
Si utilizas este documento o alguna parte de él, te agradecemos que lo cites. ¡Muchas gracias!
Clustering con Python por Joaquín Amat Rodrigo, disponible bajo una licencia Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0 DEED) en https://cienciadedatos.net/documentos/py20-clustering-con-python.html
¿Te ha gustado el artículo? Tu ayuda es importante
Tu contribución me ayudará a seguir generando contenido divulgativo gratuito. ¡Muchísimas gracias! 😊



Este documento creado por Joaquín Amat Rodrigo tiene licencia Attribution-NonCommercial-ShareAlike 4.0 International.
Se permite:
-
Compartir: copiar y redistribuir el material en cualquier medio o formato.
-
Adaptar: remezclar, transformar y crear a partir del material.
Bajo los siguientes términos:
-
Atribución: Debes otorgar el crédito adecuado, proporcionar un enlace a la licencia e indicar si se realizaron cambios. Puedes hacerlo de cualquier manera razonable, pero no de una forma que sugiera que el licenciante te respalda o respalda tu uso.
-
No-Comercial: No puedes utilizar el material para fines comerciales.
-
Compartir-Igual: Si remezclas, transformas o creas a partir del material, debes distribuir tus contribuciones bajo la misma licencia que el original.